Causes of Neuromuscular Weakness in Dogs
JP McCue, DVM, DACVIM (SAIM & Neurology), The Animal Medical Center, New York, New York
This interactive article reviews clinical features that can help identify and distinguish common causes of neuromuscular weakness.
Introduction
Localizing the cause of weakness in the neuromuscular system can be challenging. Diagnosis requires careful examination and analysis of patient history and clinical sign progression.
Neuromuscular disorders generally reflect disease of the lower motor neuron unit. Sensory function remains intact in most cases. Different diseases affecting the lower motor neuron unit can cause similar clinical signs (Table 1), including decreased muscle tone, reduced reflexes, and muscle atrophy. Autonomic signs and cranial nerve deficits can be present with generalized muscle weakness. Pattern recognition can help determine the most likely differential diagnosis, as well as appropriate diagnostic testing and treatment.
Table 1: Summary Of Clinical Signs
Case 1
Kaiser, a 4-year-old neutered male German shepherd dog, is presented for reluctance to climb stairs and a slower pace and frequent sitting during walks. Clinical signs were first observed by his owner one week prior to presentation, and he was taken to his primary clinician. CBC, serum chemistry profile, and pelvic radiography results were within normal limits. Treatment with carprofen (2 mg/kg PO every 12 hours) was initiated for possible soft tissue injury, but Kaiser did not show improvement.
On current examination, Kaiser is bright and alert. Thoracic auscultation is normal, and dorsal pedal pulses are strong and synchronous. He is reluctant to stand for examination and frequently sits or lies down. Orthopedic examination shows no pain, joint effusion, or decreased range of motion. Kaiser has good muscle condition (no atrophy), normal muscle tone, and normal spinal reflexes. Cranial nerve examination is normal. He collapses during postural reactions and hopping assessment, but postural placement tests are considered normal when his weight is supported. He can be enticed to walk for gait analysis (Video 1); steps are shortened in all limbs, body weight is shifted to the thoracic limbs, and there are slight tremors in the limbs, as with exertion. He quickly appears to weaken and lies down in a sternal position.
Examination reveals muscle weakness exacerbated by exertion. Acquired myasthenia gravis is a neuromuscular junction disease in which antibodies to nicotinic acetylcholine receptors interfere with acetylcholine binding to postsynaptic receptors on the muscle membrane. The fulminant form of this disease results in severe, acute, diffuse neuromuscular weakness accompanied by megaesophagus, but the typical presentation is exercise-induced weakness.1 Although presentation can be similar to other acute lower motor neuron diseases, a primary difference is preservation of spinal reflexes.2 Motor function deficits in postural placement testing can often be alleviated by supporting the patient’s weight.
Definitive diagnosis of myasthenia gravis is based on identification of serum antibodies against acetylcholine receptors. This test is considered extremely sensitive and specific3; however, prior administration of immunomodulatory drugs (eg, corticosteroids), low levels of circulating antibodies, and rare forms of antibodies directed against other portions of the acetylcholine receptor transmembrane signaling complex can cause false negative results.4
Edrophonium testing is often used initially to support a diagnosis; however, it is nonspecific, as other lower motor neuron diseases may respond to testing.3,4 Edrophonium is not currently available in the United States. Neostigmine is often used in place of edrophonium; however, caution should be used, as onset of action is slower by several minutes and duration of action is longer. Serologic testing remains the gold standard.
Edrophonium was available when Kaiser was presented. After administration of edrophonium IV, he is immediately able to stand and walk for ≈30 feet before becoming weak and collapsing (Video 2); this brief duration of effect is expected, and the response is consistent with myasthenia gravis, justifying treatment with a longer-acting anticholinesterase while waiting for serologic confirmation.
Although German shepherd dogs have a recognized predisposition for degenerative myelopathy, mean age of onset of clinical signs is 8 years of age,5 making Kaiser young for this diagnosis. Patients with degenerative myelopathy are classically presented with upper motor neuron paraparesis that slowly progresses over many months. Proprioceptive deficits are noted before onset of paresis. Thoracic limb paresis does not develop until late in the course of disease. A subset of patients have absent patellar reflexes.5
Although Kaiser exhibited predominant pelvic limb weakness, lumbosacral disease is unlikely considering his lack of pain on orthopedic evaluation and previously normal pelvic radiographs. Spinal radiography is a poor predictor of lumbosacral disease in dogs. Reluctance to walk and climb stairs is common in these patients, but weakness is not typical.
Tick paralysis and botulism can result in generalized lower motor neuron weakness with flaccid paralysis that is generally rapid in onset.2 Patients with botulism commonly show cranial nerve dysfunction; concurrent autonomic dysfunction is also possible.6 Kaiser did not show evidence of cranial nerve dysfunction, despite signs of generalized weakness with exercise.
Myasthenia gravis is confirmed. Acetylcholine receptor antibody titer is 2.6 nmol/L (positive, >0.6 nmol/L). Primary treatment for acquired myasthenia gravis generally involves use of a long-acting anticholinesterase, so pyridostigmine bromide is administered.
Pyridostigmine bromide (0.2-2 mg/kg PO every 8-12 hours) is typically started at the low end of the range and titrated to effect. Doses >2 mg/kg increase the risk for systemic toxicosis.7 Dose should be adjusted based on clinical response (informed by examination and signs of weakness during daily activity reported by the pet owner). Excessive use of anticholinesterase drugs increases risk for exacerbating weakness due to accumulation of acetylcholine at the neuromuscular junction. Adverse effects of ptyalism and diarrhea may also occur. Incremental dose adjustments that do not result in improved muscle strength should be lowered.
Immune suppression is controversial and should be used with caution in patients with concurrent aspiration pneumonia secondary to megaesophagus.8,9 Corticosteroids should be used with caution, as they may exacerbate muscle weakness in compromised patients. Immunosuppressant drugs may be reserved for patients with severe signs that do not respond to acetylcholinesterase inhibition.
Therapeutic plasma exchange (TPE) can be an alternative to oral medications in the acute setting. TPE has been used to treat several immune-mediated diseases in dogs, including myasthenia gravis,1,10 and has been used successfully at the author’s institution to manage dogs with moderate to severe signs of myasthenia gravis. TPE can result in rapid improvement without the potential complications or delay in effect of other immunosuppressive therapies. Early referral to a specialty center offering TPE should be considered in patients with moderate to severe signs or poor response to acetylcholinesterase therapy.
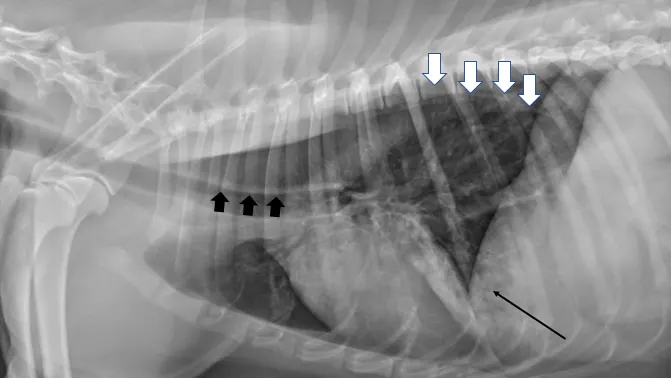
Thoracic radiography is important for diagnostic evaluation of patients with myasthenia gravis (Figure). Dogs with this disease have a high incidence of megaesophagus, as the esophagus contains skeletal muscle throughout its length. Risk for aspiration pneumonia is high and can contribute to neuromuscular weakness, impacting treatment.11

Left lateral radiograph of a dog with myasthenia gravis. The esophagus is diffusely dilated with gas (white arrows), creating a tracheal stripe sign along the dorsal margin of the trachea (wide black arrows). Focal opacity with alveolar pattern in the ventral lung field (thin black arrow) is consistent with aspiration pneumonia.
Megaesophagus has important implications for nursing care; patients should be maintained in an upright position during and after feeding. Megaesophagus may not resolve with treatment despite clinical improvement in muscle weakness.3 Supportive care in patients with esophageal dysfunction is important to avoid nutritional deficiencies and dehydration that can further compromise weakness. Construction or purchase of a chairlike device that supports the dog in a vertical position while eating can help facilitate management. Nursing care and owner education are essential for successful management.
Thoracic radiography can also be used to screen for a cranial mediastinal mass. Thymoma has been associated with myasthenia gravis and can affect case management,3,4,11 as removal may be warranted. Thymectomy in patients with thymoma-associated myasthenia gravis can improve or resolve clinical signs.12 Cats have a higher incidence of concurrent cranial mediastinal masses than dogs.3
Abdominal ultrasonography may be indicated in patients presented with ptyalism or vomiting. Careful owner questioning is important to be able to distinguish vomiting from regurgitation. Regurgitation reflects pharyngeal and/or esophageal dysfunction, which can be present in myasthenia gravis and prompt thoracic radiography.
Autonomic dysfunction is not a prominent feature of myasthenia gravis; therefore, a Schirmer tear test does not inform the initial patient evaluation.
CSF would be normal in these patients, as myasthenia gravis is a disease of the neuromuscular junction without CNS involvement.
Concurrent urinary tract disorders are not associated with myasthenic syndromes.
Case 2
Carly, a 4-year-old spayed golden retriever, is presented with a several-month history of nonspecific inactivity and decreased appetite. She was previously evaluated at another clinic. Repeat CBC and serum chemistry profiles showed persistent liver enzyme elevations (Table 2). Abdominal ultrasonography did not show abnormalities, and bile acid profile (pre- and postprandial determinations) was normal. Liver biopsy showed hepatocellular glycogen accumulation. Vector-borne tick PCR panel was negative. Because examination findings were inconclusive, Carly was referred for decreased activity and weight loss.
Table 2: Serum Chemistry Profile Results
On current examination, Carly is thin and immediately sits or lies down if made to stand. The owner reports Carly coughs while eating. Gait analysis shows short interstep distance in all limbs with weight shifted to the thoracic limbs. Fasciculation of the proximal appendicular muscles is observed when she is weight-bearing. Examination does not show localized pain. Spinal reflexes, postural reactions, and cranial nerve examination are normal. Generalized muscle atrophy is appreciated and notable in the temporalis muscle area (Video 3).
An abbreviated in-clinic serum chemistry profile reveals progressive liver enzyme (ie, ALP, ALT, AST) elevations. A more complete serum chemistry profile submitted to an external laboratory demonstrates creatine kinase elevation and further transaminase elevations. Creatine kinase had not been evaluated previously.
Polymyositis is inflammation of skeletal muscle that can result in muscle weakness and pain. Muscle fibrosis can occur following chronic polymyositis. Polymyositis is a differential diagnosis based on creatine kinase elevation and examination findings.
Creatine kinase is a cytosolic enzyme predominantly found in skeletal and cardiac muscle. Isoforms of this enzyme in the brain do not affect serum creatine kinase activities.13 Elevations in serum creatine kinase are considered specific for skeletal muscle injury because of the larger amount of skeletal muscle relative to CNS or cardiac tissues. Dysphagia, dysphonia, and regurgitation are possible because skeletal muscle is present in the entire upper GI and respiratory pathways of dogs. A focal form of myositis involving pharyngeal/laryngeal muscles may occur with dysphagia and/or dysphonia as the predominant clinical signs.
Creatine kinase has a short half-life (2-3 hours in dogs); persistently elevated values indicate ongoing muscle injury. Moderate increases in creatine kinase can be seen in young patients and all ages of athletic dogs following high levels of exertion. Prolonged recumbency and anorexia can result in significant creatine kinase elevations, particularly in cats.14
AST, a cytosolic enzyme, is a common indicator of liver dysfunction in small patients; however, it is not specific to the liver. Skeletal muscle contains the highest concentrations of AST among organ systems and is indicative of cellular injury.13 ALT, another cytosolic enzyme fairly specific to the liver in dogs, is also present in skeletal muscle.13
Ongoing transaminase elevation, particularly AST elevation in excess of ALT, indicates muscle injury and should prompt creatine kinase evaluation. Creatine kinase is a specific, sensitive marker of muscle dysfunction but does not reveal the cause of injury. Further testing is often necessary to rule out genetic, infectious, and autoimmune diseases.
Needle electromyography of the paraspinal and limb musculature in Carly revealed a generalized distribution of abnormal, spontaneous activity characterized by fibrillation potentials and positive sharp waves. Biopsy results of the triceps and vastus lateralis muscles were consistent with immune-mediated polymyositis.
Carly exhibited rapid clinical response to prednisone (1 mg/kg PO every 24 hours, tapered over 4 weeks following clinical and laboratory resolution) and cyclosporine (7 mg/kg PO every 12 hours). Combined treatment allowed a rapid reduction in glucocorticoid dose following clinical response, which helped minimize adverse effects of glucocorticoids on muscle mass and evaluation of hepatocellular enzymes.
Transaminase elevations in this patient initially focused the diagnostic investigation on the liver; however, elevations are nonspecific to cause. The pattern of liver enzyme elevation can help identify the pathologic mechanism. Vacuolar hepatopathy is most often characterized by elevations in ALP in excess of transaminase elevations; bile acids may be elevated. ALP was normal in this patient.
Significant liver enzyme elevations are not common with microvascular dysplasia, although bile acid elevations may occur. In this patient, bile acid determination after an appropriate fast and feeding were normal, as was ALP throughout repeated testing. Liver biopsy ultimately excluded microvascular dysplasia. Glycogen accumulation is a nonspecific finding and should be interpreted in combination with serum chemistry profile and clinical data. Hepatocellular glycogen accumulation was considered secondary to illness and inflammation related to myopathy.
Weakness associated with myasthenia gravis is most commonly associated with exertion (not reported in this case, although weakness can be seen in the video). Hepatocellular enzyme and creatine kinase elevation with muscle atrophy are not features of myasthenia gravis.3,4,11
Artifactual increases of creatine kinase are possible with sample handling and venipuncture, but activity rarely causes >3- to 4-fold increase above the high end of the reference range.
Case 3
Max, a 6-year-old neutered male Pomeranian, is presented for acute-onset tetraparesis that has progressed over several hours (Video 4); he was normal the evening before examination. Max is alert and responsive and appears mentally appropriate but is unable to bear weight on any limb and has diffuse weakness, flaccid muscle tone, and a hoarse bark. Spinal reflexes cannot be elicited in the pelvic limbs and are weak in the thoracic limbs. Cranial nerve examination is normal.
Severe weakness and/or paralysis in all limbs most likely indicates a neuromuscular process. A diffuse lesion of the spinal cord resulting in flaccid appendicular weakness is unlikely. Loss of spinal reflexes in all limbs further supports peripheral neuropathy. In this patient, neuropathy is predominately affecting the lower motor neurons of the limbs.
Neuromuscular disorders often show predominant motor neuron involvement. A lower motor neuron unit is composed of a neuronal cell body in the ventral gray matter of the spinal cord, its axon and axon terminus, neuromuscular junction, and muscle innervated by the neuronal axon.2 Clinically, these parts are considered a functional unit, and it can be difficult to identify which part of the unit is causing dysfunction.
The pattern of neurologic deficits can help establish neurologic localization. Sensory function remains intact in most cases, but certain disease processes can affect both motor and sensory systems.2 Autonomic dysfunction is possible with some diseases and can help prioritize differential diagnoses. For example, botulinum toxin targets the presynaptic surface of cholinergic neurons. Within the peripheral nervous system, acetylcholine is released at both the neuromuscular junction and parasympathetic synapses, resulting in muscle weakness and autonomic signs (eg, ileus, detrusor weakness, mydriasis). In contrast, myasthenia gravis weakness is caused by antibodies targeting elements of acetylcholine receptors of skeletal muscle membranes at the neuromuscular junction. Autonomic signs are uncommon. Early recognition and diagnosis of neuromuscular weakness are essential for good prognosis in patients with acute lower motor neuron weakness.
Segmental spinal lesions are unlikely in Max based on patient history and examination findings. A C6-T2 lesion would not cause flaccid weakness and loss of reflexes in pelvic limbs. Lesions of the C1-C5 and T3-L3 spinal segments result in spasticity and hyperreflexia, reflecting interruption of upper motor neuron spinal tracts.
Flaccid weakness of all limbs would be unusual for a brain or spinal lesion and would require a process affecting both the cervical and lumbar intumescences.
Differential diagnoses for Max include idiopathic polyradiculoneuritis, myasthenia gravis (fulminant form), botulism, tick paralysis, coral snake envenomation, and organophosphate toxicity.
Max has no known toxin exposure, is fed a home-prepared diet with ingredients the owners also eat, is predominantly kept indoors, and lives in an urban area but visits local parks for supervised play. He has no history of vomiting, diarrhea, or voiding dysfunction. Coral snake envenomation is a consideration in some geographic areas of the United States (eg, Southeast, Texas), but Max is located in the Northeast with no recent travel history.
Following initial examination, Max becomes weaker and begins to hypoventilate. Arterial blood gas confirms respiratory acidosis. Pulse oximetry is 88% despite supplemental oxygen, and he displays increased inspiratory effort. Cranial nerve examination remains normal. Oral examination shows no ptyalism or obstructive process, but arytenoid movement is reduced. Thoracic radiography is normal. Max is placed on mechanical ventilation due to progressive hypoventilation. CSF tap is performed with Max under a high plane of sedation, and the fluid is analyzed; results are normal.
Despite a predominantly indoor lifestyle and urban environment, Max visits local parks. He has an especially dense haircoat that precluded identification of attached ectoparasites. He rapidly deteriorated after presentation and required mechanical ventilation. Once sedated and stabilized, Max was shaved to facilitate inspection for ectoparasites. An engorged tick was found in the skinfold of his left axilla and removed, including attached mouthparts, with tweezers.
This case highlights the importance of early recognition of lower motor neuron weakness signs. These patients should be handled carefully to avoid added stress. A thorough examination for ticks should be performed in any patient presented for rapidly progressive lower motor neuron signs. Max made rapid improvement within several hours of tick removal (Video 5). He was weaned from the ventilator and discharged the following day.
There is no screening test for tick paralysis; therefore, a high index of suspicion is necessary based on clinical presentation in areas where ticks are endemic. In this case, rapid deterioration and need for ventilation made it difficult to identify the tick early during examination. Removal of attached ticks, including mouthparts buried in the skin, typically results in rapid improvement, with resolution in 24 to 72 hours. There are distinct regional differences in outcome with tick paralysis. Cases in Australia, for example, are more clinically severe than those in the United States due to differences in tick species and mechanism of toxicity (see Tick Paralysis).15,16 In Australia, clinical signs may take days to improve following tick removal, and recovery may be incomplete in some patients. These patients require extensive supportive care, can develop multiple complications, and have a guarded prognosis.
Thorough patient history is needed to rule out or distinguish toxin exposure (eg, organophosphate). Max is a small-breed dog that is usually supervised in a controlled environment, and he was not presented with autonomic signs (eg, hypersalivation, mydriasis, urine leakage); therefore, toxin exposure could be excluded.
Botulism is caused by Clostridium botulinum toxin. Ingestion of preformed toxin in carrion or contaminated meat sources is the most common form of intoxication in dogs and is unlikely in this patient.6 Lack of autonomic signs (eg, hypersalivation, mydriasis, urine leakage) also make botulism less likely.
Although fulminant myasthenia gravis can result in profound weakness and paralysis, it has a high incidence of megaesophagus and facial paralysis.11
Tick Paralysis
Tick paralysis results in severe, rapidly progressive, flaccid paralysis. Most cases are reported in the United States, Canadian Pacific Northwest, and Australia, although there is worldwide distribution. Dermacentor spp is the predominant vector in the United States. Vector-borne illness is increasingly reported throughout the United States, including in the Southeast and East, due to the expanding range of various tick species.16 Ixodes holocyclus is the predominant tick species in Australia. Peak incidence of tick paralysis occurs in the spring in Australia and the United States.15
Tick paralysis results from tick attachment and blood feeding. Onset of clinical signs can be several days after exposure. Differences in clinical severity may reflect tick species differences. The mechanism of neurotoxicity differs between Dermacentor spp and Ixodes spp.23 For Dermacentor spp, studies suggest axonal conduction is blocked by a toxic element in saliva.23 Ixodes spp produce a more severe form of paralysis via botulinum-like activity, whereby acetylcholine release by the presynaptic neuron of the neuromuscular junction is inhibited.19 Recovery is longer, and advanced supportive measures (eg, mechanical ventilation) are needed, even after the tick has been removed.
In North America, prognosis is very good with rapid identification and tick removal. Prognosis can be more guarded in Australia, although recovery is possible with rapid identification and extensive supportive care. Careful examination for ticks is indicated in patients with lower motor neuron weakness. Multiple ticks may be attached and can be difficult to locate in skin folds, within the ear canal, in the inguinal area, or between digits. Rapid recovery following removal is necessary for diagnosis.
Acute idiopathic polyradiculoneuritis is an important differential diagnosis and cannot be distinguished based on clinical signs alone. Tick paralysis and acute canine polyradiculoneuritis share several clinical features. Spinal and distal hyperesthesia is not a consistent feature of polyradiculoneuritis but, when present, helps distinguish it from other forms of neuromuscular weakness. Polyradiculoneuritis causes CSF abnormalities, differentiating it from many other causes of lower motor neuron weakness; patients can display a high protein level with normal nucleated cell count.15,20
Collection of CSF is not common in clinical evaluation of dogs with lower motor neuron weakness because of risks for poor ventilation and aspiration pneumonia. Sedation and anesthesia should be avoided when possible in patients with neuromuscular weakness. Careful handling and stress reduction are important.
Case 4
Raleigh, a 5-year-old neutered male springer spaniel, is presented for a 6-day history of progressive weakness. Clinical signs began in the pelvic limbs and slowly progressed to all limbs. On examination, Raleigh is unable to stand (Video 6). Muscle fasciculations (particularly in the pelvic limbs) can be seen while he is weight-bearing, and the thoracic limbs drag because of inability to extend the paws. Muscle tone is reduced in all limbs, but he can support his head and wag his tail. Postural reactions are absent in all limbs, even when supported. Spinal reflexes are reduced in all limbs. Palpation along the spine elicits a hyperesthestic response. Cranial nerve evaluation demonstrates mild facial paresis but is otherwise normal, and the patient exhibits normal mentation. Thoracic radiographs are normal. Serum chemistry profile (including creatine kinase), CBC, and thyroid testing results are within normal limits.
Polyradiculoneuritis is considered an immune-mediated disease with similarities to Guillain-Barré syndrome in humans.1,17,18 Immunoglobulin-mediated inflammation of the ventral nerve roots results in severe, progressive weakness of all limbs. Hyperesthesia can occur and may be due to inflammation affecting the sensory nerve roots adjacent to more severely affected ventral motor nerve rootlets.2 Polyradiculoneuritis-associated hyperesthesia is not seen with other causes of lower motor neuron weakness. Potential CSF changes are also unique; elevated protein with normal cell count (ie, albuminocytologic dissociation) is a typical pattern.19 Electrodiagnostic evaluation of the F wave to assess the proximal portion of the motor neuron can be performed by veterinary neurologists; an abnormal F wave further confirms diagnosis.17
Although polyradiculoneuritis is commonly called coonhound paralysis due to association with raccoon saliva exposure 7 to 14 days prior to onset of clinical signs in some dogs, most cases do not have this history. Association with Campylobacter spp infection has been reported, and consumption of raw chicken is a risk factor.18 Careful owner questioning, including diet history, can aid diagnosis.
Treatment for polyradiculoneuritis typically involves supportive care, with slow recovery expected over 3 to 6 months.20 Focused rehabilitative treatment is essential to recovery. Studies have shown faster recovery in veterinary patients treated early with human IV immunoglobulin.21 Therapeutic plasma exchange has emerged as an important treatment for many antibody-mediated immune diseases in dogs, is well established in humans, and may have a role in treating polyradiculoneuritis.10,22 At the author’s institution, therapeutic plasma exchange has been used successfully in patients with polyradiculoneuritis and fulminant myasthenia gravis.
Raleigh was treated with human IV immunoglobulin, which appeared to halt disease progression, and aggressive rehabilitation therapy, including an underwater treadmill and temporary use of a 4-wheeled cart. He made a complete recovery in 10 weeks (Video 7).
Loss of muscle tone and reduced spinal reflexes are unusual with myasthenia gravis.
Lack of myalgia, absent reflexes, and normal creatine kinase make polymyositis less likely.
Abnormal spinal reflexes and reduced muscle tone are indicative of lower motor neuron weakness, resulting in this patient’s inability to ambulate. Polyarthropathy does not result in these signs of weakness.
Several clinical findings can help distinguish polyradiculoneuritis from tick paralysis.20,21 Patients with polyradiculoneuritis can often wag their tails, and cranial nerve deficits are uncommon except for facial paralysis and dysphonia. Spinal hyperesthesia and sensitivity when feet are touched can occasionally be seen with polyradiculoneuritis.21