
Induction or inhibition of the hepatic and GI phase I metabolizing enzymes known as cytochrome P450s (CYP450) can result in drug-drug interactions (Figure). Induction or inhibition can be broad spectrum, with multiple CYP450 isoforms affected, or narrow spectrum, with a single isoform affected. A drug-drug interaction only occurs if the coadministered drug is metabolized by the same CYP450 isoform as the inducer or inhibitor; the likelihood of an interaction is therefore increased with broad-spectrum inducers and inhibitors. In addition, drug-metabolizing isoforms differ among species. For example, 2 drugs that interact in dogs may not interact similarly in cats. Studies should be conducted for each species to determine the effects of coadministration on metabolism.
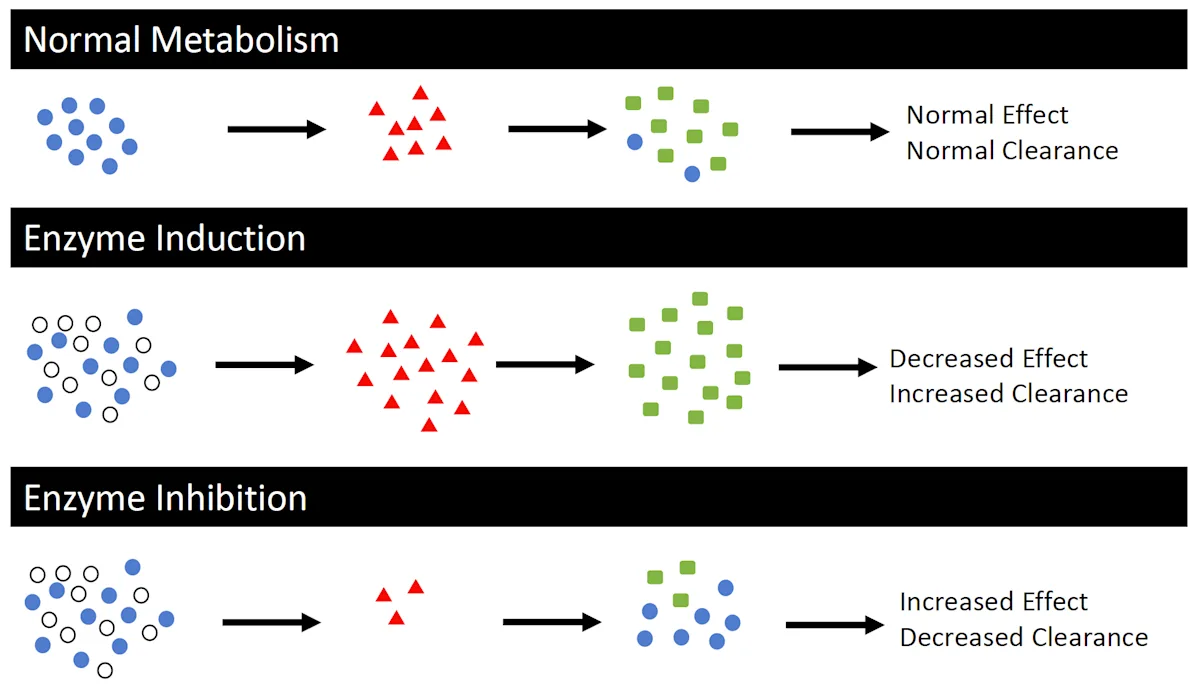
Schematic representation of normal drug metabolism when a single drug (drug 1, blue circles) is administered alone and when coadministered with a CYP450 inducer or inhibitor (white circles). The CYP450 inducer increases CYP450 enzymes (red triangles), thereby increasing production of drug 1 metabolites (green squares), which may decrease drug effect due to an increased clearance of active parent compounds. The CYP450 inhibitor decreases CYP450 enzymes, thereby decreasing production of drug 1 metabolites, which may increase drug effect due to a decreased clearance of active parent compounds. This assumes the parent compound is typically metabolized to an inactive metabolite.
Induction
Induction of CYP450 enzymes results in decreased concentration of the affected parent drug and increased concentration of its metabolites. Depending on whether the metabolites are inactive or active, drug efficacy and risk for toxicity can decrease or increase, respectively. Induction requires activation of transcription factors in the cell nuclei and exposure to multiple drug doses.
Inhibition
Inhibition of CYP450s has the opposite effect of induction, resulting in higher concentrations of the affected parent drug and lower concentrations of its metabolites. Decreased production of inactive metabolites can increase drug efficacy or toxicity due to higher concentrations of the active parent drug compound. If, however, normal metabolism results in production of active metabolites, then inhibition of metabolism may result in decreased production of those active metabolites, which would decrease drug efficacy. Inhibition of CYP450s can occur via reversible binding to the active or allosteric site on the enzyme or irreversible binding to the enzyme that forms a stable enzyme–intermediate complex. Because these mechanisms occur via binding of circulating enzymes, inhibition occurs more rapidly than induction.
Drug-Drug Interactions Resulting From CYP450 Induction
Phenobarbital
Phenobarbital is a relatively broad-spectrum inducer of drug metabolism in dogs—inducing CYP2A, CYP2B, CYP3A, and phase II uridine diphosphate–glucuronosyltransferase metabolizing enzymes—and therefore has several known drug-drug interactions.1 These interactions are clinically significant because lifelong administration of phenobarbital for treatment of seizures is common and interactions with other anticonvulsants can occur. Phenobarbital is also considered to be an autoinducer (ie, can induce its own metabolism) with chronic administration (30-90 days); higher doses (400 mg/day) induce a significant increase in clearance compared to lower doses (15 mg/day).2 A study evaluating a loading dose of 12 mg/kg PO every 24 hours, followed by 6 mg/kg PO every 24 hours for 4 weeks did not demonstrate increased clearance or shortened half-life, although the administration period may have been too short to detect a significant difference.3
Zonisamide may be administered concurrently with phenobarbital in patients that have become refractory to phenobarbital alone. In one study in dogs, administration of phenobarbital for 35 days resulted in significant decreases in the area under the curve (ie, measurement of total drug exposure over time), maximum plasma concentration, half-life, and bioavailability of zonisamide that lasted up to 10 weeks after discontinuation of phenobarbital.4 These alterations in pharmacokinetic parameters were due to increased clearance from induction of CYP2C and CYP3A enzyme subfamilies. Similar changes in the pharmacokinetics of levetiracetam have been found in dogs given levetiracetam and phenobarbital concurrently. The metabolism of levetiracetam is not dependent on CYP450s in dogs, but the mechanism of metabolism induction is thought to be due to phenobarbital-induced oxidation pathways.5 Intravenous phenobarbital may also reduce the duration of anesthesia provided by xylazine and ketamine in dogs.6 The exact mechanism of this interaction was not investigated in this study but was proposed to be through induction of CYP450s; ketamine, in particular, has been shown to be metabolized by CYP450s in dogs, horses, and humans.7
The effects of phenobarbital enzyme induction in cats are not well documented. One study found that 21 days of phenobarbital administration at therapeutic doses did not significantly alter phenobarbital pharmacokinetics.8 Another study found that phenobarbital decreased the bioavailability of cyclosporine following oral administration for 28 days.9
Rifampin
Rifampin induces hepatic and intestinal CYP3A in dogs and thus may increase clearance or absorption of other drugs metabolized through these pathways.10 A significant reduction in midazolam exposure in dogs, determined by a decrease of 7.7 times in the area under of the curve, was noted on days 9 and 11 after initiation of rifampin treatment.11 Rifampin use in small animals is rare but increasing, as this drug may be the only treatment option for methicillin-resistant Staphylococcus pseudintermedius.
Drug-Drug Interactions Resulting From CYP450 Inhibition
Azole Antifungals
The mechanism of action of azole antifungals is inhibition of the CYP450 enzyme 14 alpha-demethylase that converts lanosterol into ergosterol, a vital component of fungal cell membranes. Drug-drug interactions occur with this family of antifungals through inhibition of other CYP450s.
Fluconazole
Administration of fluconazole in dogs prior to anesthesia with midazolam and ketamine can result in a prolonged time to standing during recovery, as well as decreased clearance and increased area under the curve of midazolam and ketamine.12
Coadministration of fluconazole and tramadol in dogs can alter the metabolite profile of tramadol by decreasing production of the inactive metabolites N-desmethyltramadol (M2) and N,O-didesmethyltramadol (M5), thus increasing plasma concentrations of the parent drug and its active metabolite O-desmethyltramadol (M1).13 The clinical significance of this is unknown.
Coadministration of fluconazole and methadone can have a beneficial effect on methadone pharmacokinetics and pharmacodynamics in dogs. Oral methadone alone undergoes rapid first-pass metabolism and does not reach therapeutic blood concentrations. Addition of fluconazole results in decreased methadone clearance with an increased area under the curve and half-life and a prolonged analgesic effect comparable to injectable methadone.14
Ketoconazole
Administration of ketoconazole with midazolam and fentanyl in dogs has been shown to prolong recovery and alter the pharmacokinetics of midazolam but not fentanyl.15 In that study, ketoconazole plasma concentrations increased over 5 consecutive days of administration, indicating ketoconazole is an autoinhibitor (ie, can decrease its own metabolism over time).15
Ketoconazole has been used with cyclosporine A in dogs with atopy and allergic skin disease. In one study, coadministration reduced the metabolism of cyclosporine A, resulting in higher concentrations of cyclosporine A in the plasma and skin that allowed a 38% reduction in the cyclosporine dose and significant reduction of treatment cost.16
In cats, ketoconazole has been shown to inhibit the metabolism of midazolam in vitro and quinidine in vivo.17 Similarly, ketoconazole can decrease the clearance and increase the plasma concentration of cyclosporine A, enhancing the immunosuppressive effects of cyclosporine A and enabling once daily administration for prevention of allograft rejection following renal transplants.18
Ketoconazole is an inhibitor of p-glycoprotein efflux pumps, which may play a role in these drug interactions.
Chloramphenicol
Chloramphenicol can irreversibly bind and inhibit multiple CYP450s, resulting in strong and rapid inhibition of the metabolism of other CYP450 substrates.
Coadministration of chloramphenicol and phenobarbital can result in phenobarbital toxicosis in dogs, despite phenobarbital being a CYP450 inducer.19
Chloramphenicol interacts with several drugs in dogs. Coadministration with phenytoin can increase the half-life of phenytoin by ≈5 times.20 In a study of greyhounds, chloramphenicol increased the elimination half-life and decreased the clearance of propofol by 209% and 45%, respectively, resulting in prolonged anesthesia recovery time.21 In another study of greyhounds, previous administration of chloramphenicol resulted in an increase of 90 times in the area under the curve of oral methadone.22
Fluoroquinolones
Fluoroquinolone antibiotics are selective inhibitors of CYP1A2, but the degree of inhibition varies between drugs.
The most clinically relevant fluoroquinolone drug-drug interactions are with theophylline in dogs. Coadministration of enrofloxacin and theophylline can significantly increase the maximum plasma concentration and trough concentrations of theophylline at steady state; this interaction is likely due to CYP450 inhibition by the metabolite ciprofloxacin, as enrofloxacin itself is not a strong CYP1A2 inhibitor.23 Coadministration of marbofloxacin and theophylline can decrease theophylline clearance; one study found a significant decrease in clearance with administration of marbofloxacin at 5 mg/kg but not at 2 mg/kg.24 These interactions may result in theophylline toxicosis because of the narrow therapeutic index of theophylline, particularly in dogs with renal compromise.
Conclusion
This review of clinically relevant CYP450-mediated drug-drug interactions is not exhaustive. Continued study of commonly coadministered drugs is needed to ensure safe and effective therapy for veterinary patients.